Overview
Literature Analysis
Mouse over the terms for more detail; many indicate links which you can click for dedicated pages about the topic.
Tag cloud generated 29 August, 2019 using data from PubMed, MeSH and CancerIndex
Mutated Genes and Abnormal Protein Expression (30)
 Clicking on the Gene or Topic will take you to a separate more detailed page. Sort this list by clicking on a column heading e.g. 'Gene' or 'Topic'.
Clicking on the Gene or Topic will take you to a separate more detailed page. Sort this list by clicking on a column heading e.g. 'Gene' or 'Topic'.
| Gene | Location | Aliases | Notes | Topic | Papers |
| CTNNB1 | 3p22.1 | CTNNB, MRD19, armadillo | GWS
| -CTNNB1 and Medullobalastoma
| 66 |
| TP53 | 17p13.1 | P53, BCC7, LFS1, TRP53 | GWS
| -TP53 mutation in Medulloblastoma
| 47 |
| NTRK3 | 15q25.3 | TRKC, GP145-TrkC, gp145(trkC) | Prognostic
| -NTRK3 expression in Medulloblastoma
| 46 |
| MYC | 8q24.21 | MRTL, MYCC, c-Myc, bHLHe39 | Amplification
Overexpression
| -MYC expression and amplification in Medulloblastoma
| 39 |
| PTCH1 | 9q22.32 | PTC, BCNS, HPE7, PTC1, PTCH, NBCCS, PTCH11 | GWS
| -PTCH1 and Medulloblastoma
| 39 |
| SMO | 7q32.1 | Gx, CRJS, SMOH, FZD11 | | -SMO and Medulloblastoma
| 28 |
| MYCN | 2p24.3 | NMYC, ODED, MODED, N-myc, bHLHe37 | Amplification
| -MYCN Amplification in Medulloblastoma
| 28 |
| GLI1 | 12q13.2-q13.3 | GLI | | -GLI1 and Medulloblastoma
| 24 |
| SUFU | 10q24.32 | SUFUH, SUFUXL, PRO1280 | | -SUFU and Medulloblastoma
-SUFU germline mutations in Medulloblastoma associated with Gorlin Syndrome
| 18 |
| OTX2 | 14q22.3 | CPHD6, MCOPS5 | | -OTX2 and Medulloblastoma
| 19 |
| DDX3X | Xp11.4 | DBX, DDX3, HLP2, DDX14, CAP-Rf, MRX102 | GWS
| -DDX3X and Medulloblastoma
| 10 |
| REST | 4q12 | WT6, XBR, NRSF | | -REST and Medulloblastoma
| 9 |
| SSTR2 | 17q25.1 | | | -SSTR2 Expresssion in Medulloblastoma
| 8 |
| SMARCA4 | 19p13.2 | BRG1, CSS4, SNF2, SWI2, MRD16, RTPS2, BAF190, SNF2L4, SNF2LB, hSNF2b, BAF190A | GWS
| -SMARCA4 and Medulloblastoma
| 7 |
| NGFR | 17q21.33 | CD271, p75NTR, TNFRSF16, p75(NTR), Gp80-LNGFR | | -NGFR and Medulloblastoma
| 7 |
| MYCL | 1p34.2 | LMYC, L-Myc, MYCL1, bHLHe38 | | -MYCL1 expression in Medulloblastoma
| 7 |
| ATOH1 | 4q22.2 | ATH1, HATH1, MATH-1, bHLHa14 | | -ATOH1 and Medulloblastoma
| 7 |
| KMT2D | 12q13.12 | ALR, KMS, MLL2, MLL4, AAD10, KABUK1, TNRC21, CAGL114 | GWS
| -MLL2 (KMT2D) and Medulloblastoma
| 7 |
| PPM1D | 17q23.2 | WIP1, PP2C-DELTA | | -PPM1D and Medulloblastoma
| 6 |
| KDM6A | Xp11.3 | UTX, KABUK2, bA386N14.2 | GWS
| -KDM6A and Medulloblastma
| 6 |
| HIC1 | 17p13.3 | hic-1, ZBTB29, ZNF901 | | -HIC1 and Medulloblastoma
| 6 |
| ERBB2 | 17q12 | NEU, NGL, HER2, TKR1, CD340, HER-2, MLN 19, HER-2/neu | | -ERBB2 and Medulloblastoma
| 5 |
| DONSON | 21q22.11 | B17, MIMIS, MISSLA, C21orf60 | | -DONSON and Medulloblastoma
| 5 |
| MSI1 | 12q24 | | | -MSI1 and Medulloblastoma
| 4 |
| FUT4 | 11q21 | LeX, CD15, ELFT, FCT3A, FUTIV, SSEA-1, FUC-TIV | | -FUT4 and Medulloblastoma
| 3 |
| ERBB4 | 2q33.3-q34 | HER4, ALS19, p180erbB4 | | -ERBB4 and Medulloblastoma
| 3 |
| NEURL1 | 10q24.33 | neu, NEUR1, NEURL, RNF67, neu-1, bA416N2.1 | | -NEURL1 and Medulloblastoma
| 1 |
| CNBP | 3q21.3 | DM2, ZNF9, CNBP1, PROMM, RNF163, ZCCHC22 | | -CNBP and Medulloblastoma
| 1 |
| ZNF521 | 18q11.2 | EHZF, Evi3 | | -ZNF521 and Medulloblastoma
| 1 |
| FSTL3 | 19p13.3 | FLRG, FSRP | | -FSTL3 and Medulloblastoma
| |
Note: list is not exhaustive. Number of papers are based on searches of PubMed (click on topic title for arbitrary criteria used).
GWS - Genome/Exome Wide Study, large-scale/significant (selected):
Parsons DW et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011; 331(6016):435-9
Jones DT et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012; 488(7409):100-5
Pugh TJ et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012; 488(7409)
Molecular Subgroups of Medulloblastoma
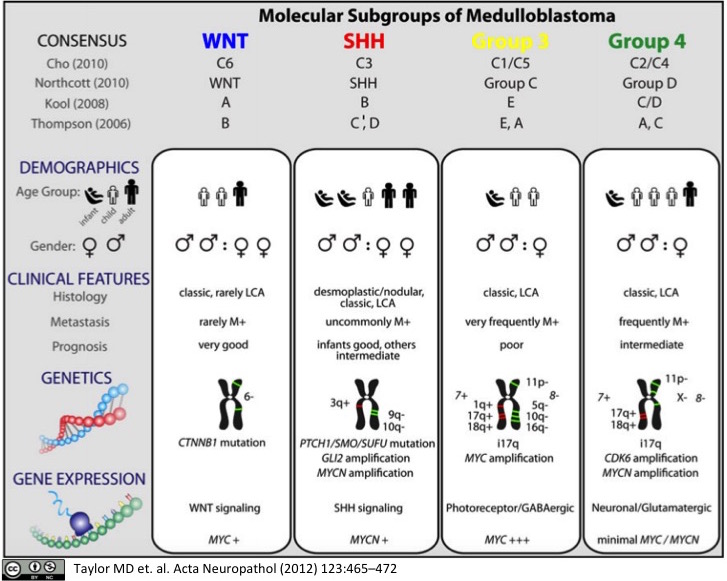
Medulloblastoma, a small blue cell malignancy of the cerebellum, is a major cause of morbidity and mortality in pediatric oncology. Current mechanisms for clinical prognostication and stratification include clinical factors (age, presence of metastases, and extent of resection) as well as histological subgrouping (classic, desmoplastic, and large cell/anaplastic histology). Transcriptional profiling studies of medulloblastoma cohorts from several research groups around the globe have suggested the existence of multiple distinct molecular subgroups that differ in their demographics, transcriptomes, somatic genetic events, and clinical outcomes. Variations in the number, composition, and nature of the subgroups between studies brought about a consensus conference in Boston in the fall of 2010. Discussants at the conference came to a consensus that the evidence supported the existence of four main subgroups of medulloblastoma (Wnt, Shh, Group 3, and Group 4). Participants outlined the demographic, transcriptional, genetic, and clinical differences between the four subgroups. While it is anticipated that the molecular classification of medulloblastoma will continue to evolve and diversify in the future as larger cohorts are studied at greater depth, herein we outline the current consensus nomenclature, and the differences between the medulloblastoma subgroups.
Kool M, Korshunov A, Remke M, et al.
Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas.Acta Neuropathol. 2012; 123(4):473-84 [
PubMed]
Free Access to Full Article Related Publications
Medulloblastoma is the most common malignant brain tumor in childhood. Molecular studies from several groups around the world demonstrated that medulloblastoma is not one disease but comprises a collection of distinct molecular subgroups. However, all these studies reported on different numbers of subgroups. The current consensus is that there are only four core subgroups, which should be termed WNT, SHH, Group 3 and Group 4. Based on this, we performed a meta-analysis of all molecular and clinical data of 550 medulloblastomas brought together from seven independent studies. All cases were analyzed by gene expression profiling and for most cases SNP or array-CGH data were available. Data are presented for all medulloblastomas together and for each subgroup separately. For validation purposes, we compared the results of this meta-analysis with another large medulloblastoma cohort (n = 402) for which subgroup information was obtained by immunohistochemistry. Results from both cohorts are highly similar and show how distinct the molecular subtypes are with respect to their transcriptome, DNA copy-number aberrations, demographics, and survival. Results from these analyses will form the basis for prospective multi-center studies and will have an impact on how the different subgroups of medulloblastoma will be treated in the future.
Pietsch T, Schmidt R, Remke M, et al.
Prognostic significance of clinical, histopathological, and molecular characteristics of medulloblastomas in the prospective HIT2000 multicenter clinical trial cohort.Acta Neuropathol. 2014; 128(1):137-49 [
PubMed]
Free Access to Full Article Related Publications
This study aimed to prospectively evaluate clinical, histopathological and molecular variables for outcome prediction in medulloblastoma patients. Patients from the HIT2000 cooperative clinical trial were prospectively enrolled based on the availability of sufficient tumor material and complete clinical information. This revealed a cohort of 184 patients (median age 7.6 years), which was randomly split at a 2:1 ratio into a training (n = 127), and a test (n = 57) dataset in order to build and test a risk score for this population. Independent validation was performed in a non-overlapping cohort (n = 83). All samples were subjected to thorough histopathological investigation, CTNNB1 mutation analysis, quantitative PCR, MLPA and FISH analyses for cytogenetic variables, and methylome analysis. By univariable analysis, clinical factors (M-stage), histopathological variables (large cell component, endothelial proliferation, synaptophysin pattern), and molecular features (chromosome 6q status, MYC amplification, subgrouping) were found to be prognostic. Molecular consensus subgrouping (WNT, SHH, Group 3, Group 4) was validated as an independent feature to stratify patients into different risk groups. When comparing methods for the identification of WNT-driven medulloblastoma, this study identified CTNNB1 sequencing and methylation profiling to most reliably identify these patients. After removing patients with particularly favorable (CTNNB1 mutation, extensive nodularity) or unfavorable (MYC amplification) markers, a risk score for the remaining "intermediate molecular risk" population dependent on age, M-stage, pattern of synaptophysin expression, and MYCN copy-number status was identified, with speckled synaptophysin expression indicating worse outcome. Test and independent validation of the score confirmed significant discrimination of patients by risk profile. Methylation subgrouping and CTNNB1 mutation status represent robust tools for the risk stratification of medulloblastoma. A simple clinico-pathological risk score was identified, which was confirmed in a test set and by independent clinical validation.
Predisposing Conditions
The vast majority of medulloblastoma cases are sporadic (non-inherited). However, a small proportion of medulloblastoma cases occur the context of hereditary syndromes which have an increased risk of cancer. Syndromes known to be associated with medulloblastoma include the following:
| Disease |
Gene(s) |
Notes |
| Basal Cell Nevus Syndrome | PTCH1 SMO PTCH2 SUFU | Basal Cell Nevus Syndrome (also known as Gorlin Syndrome) is an autosomal dominant condition characterised by the appearance of basal cell carcinomas, together with skeletal abnormalities, odontogenic keratocysts and increased risk of Medulloblastoma. Medulloblastoma develops in about 5 out of every 100 children with the syndrome. |
| Fanconi Anaemia | BRCA2 FANCD2 PALB2 FANCA FANCC more... | Fanconi Anemia (FA) is a rare autosomal recessive genetic disorder characterised clinically by progressive bone marrow failure, skeletal deformities and a predisposition to leukaemia and a wide range of cancers. Affected children usually develop severe aplastic anemia by age 8 to 9 years. |
| Li-Fraumeni syndrome | TP53 | A rare inherited autosomal dominant disorder that greatly increases the risk of developing several types of cancer, particularly in children and young adults. The most frequent types of cancer associated with Li-Fraumeni syndrome are breast cancer, osteosarcoma, and soft tissue sarcomas. People affected also have increase risk of brain tumuors, leukaemias, adrenocortical carcinoma and other types of cancer. |
| Rubinstein-Taybi Syndrome | CREBBP EP300 | Rubinstein-Taybi Syndrome (RTS) ia an autosomal dominant chromosomal disorder characterized by broad thumbs, webbing of fingers and toes, mental retardation, beaked nose, short upper lip, pouting lower lip. Individuals with RTS have an increased risk of brain tumors and occasionally other tumours. Approximately 5 % of RTS patients develop a malignancy or a benign tumor. |
| Turcot Syndrome | PMS2 MLH1 APC MSH2 MSH6 | Turcot Syndrome is characterised by malignant tumors of the central nervous system (mostly astrocytomas and medulloblastoma) associated with familial polyposis of the colon. There are different sub-types (Paraf F et al, 1997). |
BACKGROUND: Solid tumours are less oxygenated than normal tissues. This is called tumour hypoxia and leads to resistance to radiotherapy and chemotherapy. The molecular mechanisms underlying such resistance have been investigated in a range of tumour types, including the adult brain tumours glioblastoma, yet little is known for paediatric brain tumours. Medulloblastoma (MB) is the most common malignant brain tumour in children. We aimed to elucidate the impact of hypoxia on the sensitivity of MB cells to chemo- and radiotherapy.
METHODS: We used two MB cell line (D283-MED and MEB-Med8A) and a widely used glioblastoma cell line (U87MG) for comparison. We applied a range of molecular and cellular techniques to measure cell survival, cell cycle progression, protein expression and DNA damage combined with a transcriptomic micro-array approach in D283-MED cells, for global gene expression analysis in acute and chronic hypoxic conditions.
RESULTS: In D283-MED and U87MG, chronic hypoxia (5 days), but not acute hypoxia (24 h) induced resistance to chemotherapy and X-ray irradiation. This acquired resistance upon chronic hypoxia was present but less pronounced in MEB-Med8A cells. Using transcriptomic analysis in D283-MED cells, we found a large transcriptional remodelling upon long term hypoxia, in particular the expression of a number of genes involved in detection and repair of double strand breaks (DSB) was altered. The levels of Nibrin (NBN) and MRE11, members of the MRN complex (MRE11/Rad50/NBN) responsible for DSB recognition, were significantly down-regulated. This was associated with a reduction of Ataxia Telangiectasia Mutated (ATM) activation by etoposide, indicating a profound dampening of the DNA damage signalling in hypoxic conditions. As a consequence, p53 activation by etoposide was reduced, and cell survival enhanced. Whilst U87MG shared the same dampened p53 activity, upon chemotherapeutic drug treatment in chronic hypoxic conditions, these cells used a different mechanism, independent of the DNA damage pathway.
CONCLUSION: Together our results demonstrate a new mechanism explaining hypoxia-induced resistance involving the alteration of the response to DSB in D283-MED cells, but also highlight the cell type to cell type diversity and the necessity to take into account the differing tumour genetic make-up when considering re-sensitisation therapeutic protocols.
Introduction: One of the major challenges in cancer treatment is the lack of specific and accurate treatment in
cancer. Data analysis can help to understand the underlying molecular mechanism that leads to better treatment.
Increasing availability and reliability of DNA microarray data leads to increase the use of these data in a variety of
cancers. This study aimed at applying and evaluating microarray data analyzing, identification of important pathways
and gene network for medulloblastoma patients to improve treatment approaches especially target therapy. Methods:
In the current study, Microarray gene expression data (GSE50161) were extracted from Geo datasets and then analyzed
by the affylmGUI package to predict and investigate upregulated and downregulated genes in medulloblastoma. Then,
the important pathways were determined by using software and gene enrichment analyses. Pathways visualization
and network analyses were performed by Cytoscape. Results: A total number of 249 differentially expressed genes
(DEGs) were identified in medulloblastoma compared to normal samples. Cell cycle, p53, and FoxO signaling pathways
were indicated in medulloblastoma, and CDK1, CCNB1, CDK2, and WEE1 were identified as some of the important
genes in the medulloblastoma. Conclusion: Identification of critical and specific pathway in any disease, in our case
medulloblastoma, can lead us to better clinical management and accurate treatment and target therapy.
Oyefiade A, Erdman L, Goldenberg A, et al.
PPAR and GST polymorphisms may predict changes in intellectual functioning in medulloblastoma survivors.J Neurooncol. 2019; 142(1):39-48 [
PubMed]
Related Publications
PURPOSE: Advances in the treatment of pediatric medulloblastoma have led to improved survival rates, though treatment-related toxicity leaves children with significant long-term deficits. There is significant variability in the cognitive outcome of medulloblastoma survivors, and it has been suggested that this variability may be attributable to genetic factors. The aim of this study was to explore the contributions of single nucleotide polymorphisms (SNPs) in two genes, peroxisome proliferator activated receptor (PPAR) and glutathione-S-transferase (GST), to changes in general intellectual functioning in medulloblastoma survivors.
METHODS: Patients (n = 44, mean
RESULTS: We identified age at diagnosis, radiation therapy, chemotherapy, and eight SNPs associated with PPARs as predictors of general intellectual functioning. Of the eight SNPs identified, PPARα (rs6008197), PPARγ (rs13306747), and PPARδ (rs3734254) were most significantly associated with long-term changes in general intellectual functioning in medulloblastoma survivors.
CONCLUSIONS: PPAR polymorphisms may predict intellectual outcome changes in children treated for medulloblastoma. Importantly, emerging evidence suggests that PPAR agonists may provide an opportunity to minimize the effects of treatment-related cognitive sequelae in these children.
Tanori M, Pannicelli A, Pasquali E, et al.
Cancer risk from low dose radiation in Ptch1DNA Repair (Amst). 2019; 74:70-79 [
PubMed]
Related Publications
DSBs are harmful lesions produced through endogenous metabolism or by exogenous agents such as ionizing radiation, that can trigger genomic rearrangements. We have recently shown that exposure to 2 Gy of X-rays has opposite effects on the induction of Shh-dependent MB in NHEJ- and HR-deficient Ptch1
Łastowska M, Trubicka J, Karkucińska-Więckowska A, et al.
Immunohistochemical detection of ALK protein identifies APC mutated medulloblastoma and differentiates the WNT-activated medulloblastoma from other types of posterior fossa childhood tumors.Brain Tumor Pathol. 2019; 36(1):1-6 [
PubMed]
Free Access to Full Article Related Publications
Expression of the ALK gene strongly correlates with the WNT-activated medulloblastomas, which are routinely identified by detection of CTNNB1 mutation. However, some tumors have mutations in other than CTNNB1 genes. Therefore, we investigated if ALK expression may identify WNT-activated tumors without CTNNB1 mutation. In addition, we examined if ALK expression may differentiate WNT-activated medulloblastoma from other malignant posterior fossa tumors. ALK expression was analyzed using immunohistochemistry (clone D5F3) in 70 patients with posterior fossa tumours. Among 55 medulloblastomas, 6 tumors showed ALK expression in > 50% of tumor cells. In one tumor, with ALK positive reaction, negative nuclear reaction against β-catenin and the lack of CTNNB1 mutation, next generation sequencing revealed a presence of pathogenic variant c.3366_3369del in the APC gene, with homozygous deletion leading to inactivation of both copies in tumor cells. MLPA analysis displayed the presence of chromosome 6 monosomy, therefore, confirming the WNT type of this tumor. All analyzed 19 anaplastic ependymomas, 4 choroid plexus carcinomas and 2 atypical teratoid rhabdoid tumors were immunonegative for ALK expression. Therefore, we propose, that immunohistochemical detection of ALK protein should be highly recommended in routine investigation, in parallel to already established methods for identification and differentiation of WNT-activated medulloblastoma.
The aberrant activation of hedgehog (HH) signaling is a leading cause of the development of medulloblastoma, a pediatric tumor of the cerebellum. The FDA‑approved HH inhibitor, Vismodegib, which targets the transmembrane transducer SMO, has shown limited efficacy in patients with medulloblastoma, due to compensatory mechanisms that maintain an active HH‑GLI signaling status. Thus, the identification of novel actionable mechanisms, directly affecting the activity of the HH‑regulated GLI transcription factors is an important goal for these malignancies. In this study, using gene expression and reporter assays, combined with biochemical and cellular analyses, we demonstrate that mitogen‑activated kinase kinase kinase 1 (MEKK1), the most upstream kinase of the mitogen‑activated protein kinase (MAPK) phosphorylation modules, suppresses HH signaling by associating and phosphorylating GLI1, the most potent HH‑regulated transcription factor. Phosphorylation occurred at multiple residues in the C‑terminal region of GLI1 and was followed by an increased association with the cytoplasmic proteins 14‑3‑3. Of note, the enforced expression of MEKK1 or the exposure of medulloblastoma cells to the MEKK1 activator, Nocodazole, resulted in a marked inhibitory effect on GLI1 activity and tumor cell proliferation and viability. Taken together, the results of this study shed light on a novel regulatory mechanism of HH signaling, with potentially relevant implications in cancer therapy.
Goschzik T, Schwalbe EC, Hicks D, et al.
Prognostic effect of whole chromosomal aberration signatures in standard-risk, non-WNT/non-SHH medulloblastoma: a retrospective, molecular analysis of the HIT-SIOP PNET 4 trial.Lancet Oncol. 2018; 19(12):1602-1616 [
PubMed]
Free Access to Full Article Related Publications
BACKGROUND: Most children with medulloblastoma fall within the standard-risk clinical disease group defined by absence of high-risk features (metastatic disease, large-cell/anaplastic histology, and MYC amplification), which includes 50-60% of patients and has a 5-year event-free survival of 75-85%. Within standard-risk medulloblastoma, patients in the WNT subgroup are established as having a favourable prognosis; however, outcome prediction for the remaining majority of patients is imprecise. We sought to identify novel prognostic biomarkers to enable improved risk-adapted therapies.
METHODS: The HIT-SIOP PNET 4 trial recruited 338 patients aged 4-21 years with medulloblastoma between Jan 1, 2001, and Dec 31, 2006, in 120 treatment institutions in seven European countries to investigate hyperfractionated radiotherapy versus standard radiotherapy. In this retrospective analysis, we assessed the remaining tumour samples from patients in the HIT-SIOP PNET 4 trial (n=136). We assessed the clinical behaviour of the molecularly defined WNT and SHH subgroups, and identified novel independent prognostic markers and models for standard-risk patients with non-WNT/non-SHH disease. Because of the scarcity and low quality of available genomic material, we used a mass spectrometry-minimal methylation classifier assay (MS-MIMIC) to assess methylation subgroup and a molecular inversion probe array to detect genome-wide copy number aberrations. Prognostic biomarkers and models identified were validated in an independent, demographically matched cohort (n=70) of medulloblastoma patients with non-WNT/non-SHH standard-risk disease treated with conventional therapies (maximal surgical resection followed by adjuvant craniospinal irradiation [all patients] and chemotherapy [65 of 70 patients], at UK Children's Cancer and Leukaemia Group and European Society for Paediatric Oncology (SIOPE) associated treatment centres between 1990 and 2014. These samples were analysed by Illumina 450k DNA methylation microarray. HIT-SIOP PNET 4 is registered with ClinicalTrials.gov, number NCT01351870.
FINDINGS: We analysed methylation subgroup, genome-wide copy number aberrations, and mutational features in 136 assessable tumour samples from the HIT-SIOP PNET 4 cohort, representing 40% of the 338 patients in the trial cohort. This cohort of 136 samples consisted of 28 (21%) classified as WNT, 17 (13%) as SHH, and 91 (67%) as non-WNT/non-SHH (we considered Group3 and Group4 medulloblastoma together in our analysis because of their similar molecular and clinical features). Favourable outcomes for WNT tumours were confirmed in patients younger than 16 years, and all relapse events in SHH (four [24%] of 17) occurred in patients with TP53 mutation (TP53
INTERPRETATION: We define a whole chromosomal signature that allows the assignment of non-WNT/non-SHH medulloblastoma patients normally classified as standard-risk into favourable-risk and high-risk categories. In addition to patients younger than 16 years with WNT tumours, patients with non-WNT/non-SHH tumours with our defined whole chromosomal aberration signature and patients with SHH-TP53
FUNDING: Cancer Research UK, Swedish Childhood Cancer Foundation, French Ministry of Health/French National Cancer Institute, and the German Children's Cancer Foundation.
Despite improvements in overall survival, only a modest percentage of patients survives high-risk medulloblastoma. The devastating side effects of radiation and chemotherapy substantially reduce quality of life for surviving patients. Here, using genomic screens, we identified miR-584-5p as a potent therapeutic adjuvant that potentiates medulloblastoma to radiation and vincristine. MiR-584-5p inhibited medulloblastoma growth and prolonged survival of mice in pre-clinical tumor models. MiR-584-5p overexpression caused cell cycle arrest, DNA damage, and spindle defects in medulloblastoma cells. MiR-584-5p mediated its tumor suppressor and therapy-sensitizing effects by targeting HDAC1 and eIF4E3. MiR-584-5p overexpression or HDAC1/eIF4E3 silencing inhibited medulloblastoma stem cell self-renewal without affecting neural stem cell growth. In medulloblastoma patients, reduced expression of miR-584-5p correlated with increased levels of HDAC1/eIF4E3. These findings identify a previously undefined role for miR-584-5p/HDAC1/eIF4E3 in regulating DNA repair, microtubule dynamics, and stemness in medulloblastoma and set the stage for a new way to treat medulloblastoma using miR-584-5p.
Frappaz D, Faure-Conter C, Meyronet D, et al.
Are molecular subgroups of medulloblastomas really prognostic?Curr Opin Neurol. 2018; 31(6):747-751 [
PubMed]
Related Publications
PURPOSE OF REVIEW: Medulloblastoma is no more a unique disease. Clinical and biologic classification used so far are challenged by molecular classification(s). Following the consensus article that described four molecular groups of medulloblastoma in 2012, several articles in 2017 provided more relevant classifications that may impact on further clinical trial design.
RECENT FINDINGS: Though wingless (WNT) and sonic hedgehog (SHH) are defined by the activation of their respective pathways, the age and type of activation define various subgroups with specific features and outcome. Groups 3 and 4 remain ill defined. The whole population of medulloblastoma may be divided in 12 subgroups: WNTαβ, SHHαβγδ, group 3αβγ and group 4αβγ. The paediatric population may be divided in seven subgroups: WNT, SHH of infants and children, and low-risk and high-risk groups 3 and 4. SHH of infants may be divided as iSHH-I vs. iSHH-II that have different prognosis. Moreover, specific drivers of groups 3 and 4 were reported.
SUMMARY: These findings have and will have direct implications on the conception of clinical trials. Low-risk groups will benefit from less toxic therapies, and high-risk groups will benefit from targeted therapies.
Medulloblastoma is the most common malignant brain tumor of childhood. Group 3 medulloblastoma, the most aggressive molecular subtype, frequently disseminates through the leptomeningeal cerebral spinal fluid (CSF) spaces in the brain and spinal cord. The mechanism of dissemination through the CSF remains poorly understood, and the molecular pathways involved in medulloblastoma metastasis and self-renewal are largely unknown. Here we show that NOTCH1 signaling pathway regulates both the initiation of metastasis and the self-renewal of medulloblastoma. We identify a mechanism in which NOTCH1 activates BMI1 through the activation of TWIST1. NOTCH1 expression and activity are directly related to medulloblastoma metastasis and decreased survival rate of tumor-bearing mice. Finally, medulloblastoma-bearing mice intrathecally treated with anti-NRR1, a NOTCH1 blocking antibody, present lower frequency of spinal metastasis and higher survival rate. These findings identify NOTCH1 as a pivotal driver of Group 3 medulloblastoma metastasis and self-renewal, supporting the development of therapies targeting this pathway.
Kim J, Kim J, Lee Y
DNA polymerase β deficiency in the p53 null cerebellum leads to medulloblastoma formation.Biochem Biophys Res Commun. 2018; 505(2):548-553 [
PubMed]
Related Publications
Defects in DNA damage response or repair mechanisms during neurogenesis result in genomic instability, which is causative for several neural defects. These include brain tumors, particularly medulloblastoma, which occurs in the cerebellum with a high incidence in children. We generated an animal model with defective base excision repair during brain development through selective inactivation of DNA polymerase β (Polb) in neuroprogenitor cells. All of Polb conditional knockout mice developed medulloblastoma in a p53 null background, similar to the Xrcc1 and p53 double deficient animal model. XRCC1 is a scaffolding protein which is involved in DNA damage repair and binds to POLB. In both animal models, the histopathological characteristics of the medulloblastoma were similar to those of human classic medulloblastoma. Brain tumor development was slower in the Polb and p53 double null animals than in the Xrcc1 and p53 double knockout animals. Molecular marker analysis suggested that Polb- and Xrcc1-deficient medulloblastomas belonged to the SHHα subtype, underscoring the important role of genomic stability in preventing this devastating pediatric cerebellar tumor.
Leal LF, Evangelista AF, de Paula FE, et al.
Reproducibility of the NanoString 22-gene molecular subgroup assay for improved prognostic prediction of medulloblastoma.Neuropathology. 2018; 38(5):475-483 [
PubMed]
Related Publications
Medulloblastoma is the most frequent malignant brain tumor in children. Four medulloblastoma molecular subgroups, MB
Molecular classification has improved the knowledge of medulloblastoma (MB), the most common malignant brain tumour in children, however current treatments cause severe side effects in patients. Cancer stem cells (CSCs) have been described in MB and represent a sub population characterised by self-renewal and the ability to generate tumour cells, thus representing the reservoir of the tumour. To investigate molecular pathways that characterise this sub population, we isolated CSCs from Sonic Hedgehog Medulloblastoma (SHH MB) arisen in Patched 1 (
Nör C, Ramaswamy V
Clinical and pre-clinical utility of genomics in medulloblastoma.Expert Rev Neurother. 2018; 18(8):633-647 [
PubMed]
Related Publications
INTRODUCTION: Integrated genomics has significantly advanced our understanding of medulloblastoma heterogeneity. It is now clear that it actually comprises at least four distinct molecular subgroups termed Wnt/Wingless (WNT), Sonic Hedgehog (SHH), Group 3, and Group 4 with stark clinical and biological differences. Areas covered: This paper reviews advances in the classification and risk stratification of medulloblastoma, specifically integrating subgroup with clinical and cytogenetic risk factors, with a summary of the potential to lead to more precise therapies. Moreover, the current state of preclinical modeling is summarized with respect to their utility in generating new treatments and correlation with genomic discoveries. Opportunities and challenges in developing new treatment paradigms are summarized and discussed, specifically new therapies for very high-risk metastatic/MYC-amplified Group 3 and TP53-mutant SHH and reductions in therapy for lower risk groups. Expert commentary: Survival across medulloblastoma has been stagnant for over 30 years, and new treatment paradigms are urgently required. Current therapy significantly over treats a high proportion of patients leaving them with lifelong side effects; while many patients still succumb to their disease. Applying biological advances could improve quality of life for a significant proportion of patients while offering new upfront approaches to the highest risk patients.
Genomic characterization has begun to redefine diagnostic classifications of cancers. However, it remains a challenge to infer disease phenotypes from genomic alterations alone. To help realize the promise of genomics, we have performed a quantitative proteomics investigation using Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) and 41 tissue samples spanning the 4 genomically based subgroups of medulloblastoma and control cerebellum. We have identified and quantitated thousands of proteins across these groups and find that we are able to recapitulate the genomic subgroups based upon subgroup restricted and differentially abundant proteins while also identifying subgroup specific protein isoforms. Integrating our proteomic measurements with genomic data, we calculate a poor correlation between mRNA and protein abundance. Using EPIC 850 k methylation array data on the same tissues, we also investigate the influence of copy number alterations and DNA methylation on the proteome in an attempt to characterize the impact of these genetic features on the proteome. Reciprocally, we are able to use the proteome to identify which genomic alterations result in altered protein abundance and thus are most likely to impact biology. Finally, we are able to assemble protein-based pathways yielding potential avenues for clinical intervention. From these, we validate the EIF4F cap-dependent translation pathway as a novel druggable pathway in medulloblastoma. Thus, quantitative proteomics complements genomic platforms to yield a more complete understanding of functional tumor biology and identify novel therapeutic targets for medulloblastoma.
INTRODUCTION: Medulloblastoma is an aggressive but potentially curable central nervous system tumor that remains a treatment challenge. Analysis of therapeutic targets can provide opportunities for the selection of agents.
METHODS: Using multiplatform analysis, 36 medulloblastomas were extensively profiled from 2009 to 2015. Immunohistochemistry, next generation sequencing, chromogenic in situ hybridization, and fluorescence in situ hybridization were used to identify overexpressed proteins, immune checkpoint expression, mutations, tumor mutational load, and gene amplifications.
RESULTS: High expression of MRP1 (89%, 8/9 tumors), TUBB3 (86%, 18/21 tumors), PTEN (85%, 28/33 tumors), TOP2A (84%, 26/31 tumors), thymidylate synthase (TS; 80%, 24/30 tumors), RRM1 (71%, 15/21 tumors), and TOP1 (63%, 19/30 tumors) were found in medulloblastoma. TOP1 was found to be enriched in metastatic tumors (90%; 9/10) relative to posterior fossa cases (50%; 10/20) (p = 0.0485, Fisher exact test), and there was a positive correlation between TOP2A and TOP1 expression (p = 0.0472). PD-1 + T cell tumor infiltration was rare, PD-L1 tumor expression was uncommon, and TML was low, indicating that immune checkpoint inhibitors as a monotherapy should not necessarily be prioritized for therapeutic consideration based on biomarker expression. Gene amplifications such as those of Her2 or EGFR were not found. Several unique mutations were identified, but their rarity indicates large-scale screening efforts would be necessary to identify sufficient patients for clinical trial inclusion.
CONCLUSIONS: Therapeutics are available for several of the frequently expressed targets, providing a justification for their consideration in the setting of medulloblastoma.
Super-enhancers are large clusters of enhancers that activate gene expression. Broad trimethyl histone H3 lysine 4 (H3K4me3) often defines active tumor suppressor genes. However, how these epigenomic signatures are regulated for tumor suppression is little understood. Here we show that brain-specific knockout of the H3K4 methyltransferase MLL4 (a COMPASS-like enzyme, also known as KMT2D) in mice spontaneously induces medulloblastoma. Mll4 loss upregulates oncogenic Ras and Notch pathways while downregulating neuronal gene expression programs. MLL4 enhances DNMT3A-catalyzed DNA methylation and SIRT1/BCL6-mediated H4K16 deacetylation, which antagonize expression of Ras activators and Notch pathway components, respectively. Notably, Mll4 loss downregulates tumor suppressor genes (e.g., Dnmt3a and Bcl6) by diminishing broad H3K4me3 and super-enhancers and also causes widespread impairment of these epigenomic signatures during medulloblastoma genesis. These findings suggest an anti-tumor role for super-enhancers and provide a unique tumor-suppressive mechanism in which MLL4 is necessary to maintain broad H3K4me3 and super-enhancers at tumor suppressor genes.
Robinson GW, Rudneva VA, Buchhalter I, et al.
Risk-adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial.Lancet Oncol. 2018; 19(6):768-784 [
PubMed]
Free Access to Full Article Related Publications
BACKGROUND: Young children with medulloblastoma have a poor overall survival compared with older children, due to use of radiation-sparing therapy in young children. Radiotherapy is omitted or reduced in these young patients to spare them from debilitating long-term side-effects. We aimed to estimate event-free survival and define the molecular characteristics associated with progression-free survival in young patients with medulloblastoma using a risk-stratified treatment strategy designed to defer, reduce, or delay radiation exposure.
METHODS: In this multicentre, phase 2 trial, we enrolled children younger than 3 years with newly diagnosed medulloblastoma at six centres in the USA and Australia. Children aged 3-5 years with newly diagnosed, non-metastatic medulloblastoma without any high-risk features were also eligible. Eligible patients were required to start therapy within 31 days from definitive surgery, had a Lansky performance score of at least 30, and did not receive previous radiotherapy or chemotherapy. Patients were stratified postoperatively by clinical and histological criteria into low-risk, intermediate-risk, and high-risk treatment groups. All patients received identical induction chemotherapy (methotrexate, vincristine, cisplatin, and cyclophosphamide), with high-risk patients also receiving an additional five doses of vinblastine. Induction was followed by risk-adapted consolidation therapy: low-risk patients received cyclophosphamide (1500 mg/m
FINDINGS: Between Nov 27, 2007, and April 19, 2017, we enrolled 81 patients with histologically confirmed medulloblastoma. Accrual to the low-risk group was suspended after an interim analysis on Dec 2, 2015, when the 1-year event-free survival was estimated to be below the stopping rule boundary. After a median follow-up of 5·5 years (IQR 2·7-7·3), 5-year event-free survival was 31·3% (95% CI 19·3-43·3) for the whole cohort, 55·3% (95% CI 33·3-77·3) in the low-risk cohort (n=23) versus 24·6% (3·6-45·6) in the intermediate-risk cohort (n=32; hazard ratio 2·50, 95% CI 1·19-5·27; p=0·016) and 16·7% (3·4-30·0) in the high-risk cohort (n=26; 3·55, 1·66-7·59; p=0·0011; overall p=0·0021). 5-year progression-free survival by methylation subgroup was 51·1% (95% CI 34·6-67·6) in the sonic hedgehog (SHH) subgroup (n=42), 8·3% (95% CI 0·0-24·0%) in the group 3 subgroup (n=24), and 13·3% (95% CI 0·0-37·6%) in the group 4 subgroup (n=10). Within the SHH subgroup, two distinct methylation subtypes were identified and named iSHH-I and iSHH-II. 5-year progression-free survival was 27·8% (95% CI 9·0-46·6; n=21) for iSHH-I and 75·4% (55·0-95·8; n=21) for iSHH-II. The most common adverse events were grade 3-4 febrile neutropenia (48 patients [59%]), neutropenia (21 [26%]), infection with neutropenia (20 [25%]), leucopenia (15 [19%]), vomiting (15 [19%]), and anorexia (13 [16%]). No treatment-related deaths occurred.
INTERPRETATION: The risk-adapted approach did not improve event-free survival in young children with medulloblastoma. However, the methylation subgroup analyses showed that the SHH subgroup had improved progression-free survival compared with the group 3 subgroup. Moreover, within the SHH subgroup, the iSHH-II subtype had improved progression-free survival in the absence of radiation, intraventricular chemotherapy, or high-dose chemotherapy compared with the iSHH-I subtype. These findings support the development of a molecularly driven, risk-adapted, treatment approach in future trials in young children with medulloblastoma.
FUNDING: American Lebanese Syrian Associated Charities, St Jude Children's Research Hospital, NCI Cancer Center, Alexander and Margaret Stewart Trust, Sontag Foundation, and American Association for Cancer Research.
Waszak SM, Northcott PA, Buchhalter I, et al.
Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort.Lancet Oncol. 2018; 19(6):785-798 [
PubMed]
Free Access to Full Article Related Publications
BACKGROUND: Medulloblastoma is associated with rare hereditary cancer predisposition syndromes; however, consensus medulloblastoma predisposition genes have not been defined and screening guidelines for genetic counselling and testing for paediatric patients are not available. We aimed to assess and define these genes to provide evidence for future screening guidelines.
METHODS: In this international, multicentre study, we analysed patients with medulloblastoma from retrospective cohorts (International Cancer Genome Consortium [ICGC] PedBrain, Medulloblastoma Advanced Genomics International Consortium [MAGIC], and the CEFALO series) and from prospective cohorts from four clinical studies (SJMB03, SJMB12, SJYC07, and I-HIT-MED). Whole-genome sequences and exome sequences from blood and tumour samples were analysed for rare damaging germline mutations in cancer predisposition genes. DNA methylation profiling was done to determine consensus molecular subgroups: WNT (MB
FINDINGS: We included a total of 1022 patients with medulloblastoma from the retrospective cohorts (n=673) and the four prospective studies (n=349), from whom blood samples (n=1022) and tumour samples (n=800) were analysed for germline mutations in 110 cancer predisposition genes. In our rare variant burden analysis, we compared these against 53 105 sequenced controls from ExAC and identified APC, BRCA2, PALB2, PTCH1, SUFU, and TP53 as consensus medulloblastoma predisposition genes according to our rare variant burden analysis and estimated that germline mutations accounted for 6% of medulloblastoma diagnoses in the retrospective cohort. The prevalence of genetic predispositions differed between molecular subgroups in the retrospective cohort and was highest for patients in the MB
INTERPRETATION: Genetic counselling and testing should be used as a standard-of-care procedure in patients with MB
FUNDING: German Cancer Aid; German Federal Ministry of Education and Research; German Childhood Cancer Foundation (Deutsche Kinderkrebsstiftung); European Research Council; National Institutes of Health; Canadian Institutes for Health Research; German Cancer Research Center; St Jude Comprehensive Cancer Center; American Lebanese Syrian Associated Charities; Swiss National Science Foundation; European Molecular Biology Organization; Cancer Research UK; Hertie Foundation; Alexander and Margaret Stewart Trust; V Foundation for Cancer Research; Sontag Foundation; Musicians Against Childhood Cancer; BC Cancer Foundation; Swedish Council for Health, Working Life and Welfare; Swedish Research Council; Swedish Cancer Society; the Swedish Radiation Protection Authority; Danish Strategic Research Council; Swiss Federal Office of Public Health; Swiss Research Foundation on Mobile Communication; Masaryk University; Ministry of Health of the Czech Republic; Research Council of Norway; Genome Canada; Genome BC; Terry Fox Research Institute; Ontario Institute for Cancer Research; Pediatric Oncology Group of Ontario; The Family of Kathleen Lorette and the Clark H Smith Brain Tumour Centre; Montreal Children's Hospital Foundation; The Hospital for Sick Children: Sonia and Arthur Labatt Brain Tumour Research Centre, Chief of Research Fund, Cancer Genetics Program, Garron Family Cancer Centre, MDT's Garron Family Endowment; BC Childhood Cancer Parents Association; Cure Search Foundation; Pediatric Brain Tumor Foundation; Brainchild; and the Government of Ontario.
Wang J, Garancher A, Ramaswamy V, Wechsler-Reya RJ
Medulloblastoma: From Molecular Subgroups to Molecular Targeted Therapies.Annu Rev Neurosci. 2018; 41:207-232 [
PubMed]
Related Publications
Brain tumors are the leading cause of cancer-related death in children, and medulloblastoma (MB) is the most common malignant pediatric brain tumor. Advances in surgery, radiation, and chemotherapy have improved the survival of MB patients. But despite these advances, 25-30% of patients still die from the disease, and survivors suffer severe long-term side effects from the aggressive therapies they receive. Although MB is often considered a single disease, molecular profiling has revealed a significant degree of heterogeneity, and there is a growing consensus that MB consists of multiple subgroups with distinct driver mutations, cells of origin, and prognosis. Here, we review recent progress in MB research, with a focus on the genes and pathways that drive tumorigenesis, the animal models that have been developed to study tumor biology, and the advances in conventional and targeted therapy.
Zhao C, Chen Q, Li C, et al.
The association of NF2 (neurofibromin 2) gene polymorphism and the risk of medulloblastomas.Neurol Sci. 2018; 39(7):1175-1183 [
PubMed]
Related Publications
To explore the relationship between NF2 promoter gene mutation and the risk of medulloblastomas (MBs). We collected tissues from 16 MB patients and 7 age-matched non-MB controls. Gene sequencing, qPCR (real-time quantitative polymerase chain reaction), IHC (immunohistochemistry), and WB (Western blot) were used to analyze the changes in the NF2 gene sequence and expression between patients and controls. We found that NF2 promoter gene mutations occurred in MB patients. The NF2 mRNA expression was higher in the controls than in patients (p = 0.03 < 0.05); however, the results of IHC and WB demonstrated that the NF2 protein expression was significantly higher in patients than in the controls (IHC: p = 0.0001; WB: p = 0.01). There was no significant difference in the CRL4 mRNA and protein levels. In addition, NF2 protein was mainly expressed in the nucleus in MB patients, while the NF2 protein was mainly expressed in the cytoplasm in the controls. NF2 promoter mutations exist in MB patients. NF2 mRNA expression was higher in controls than patients; whereas NF2 protein level was higher in patients than in controls.
Durmaz CD, Evans G, Smith MJ, et al.
A Novel PTCH1 Frameshift Mutation Leading to Nevoid Basal Cell Carcinoma Syndrome.Cytogenet Genome Res. 2018; 154(2):57-61 [
PubMed]
Related Publications
Nevoid basal cell carcinoma syndrome (NBCCS), also known as Gorlin syndrome, is a rare multisystemic autosomal dominant disorder typically presenting with cutaneous basal cell carcinomas, multiple keratocysts, and skeletal anomalies. NBCCS is caused by heterozygous mutations in the PTCH1 gene in chromosome 9q22, in the PTCH2 gene in 1p34, or the SUFU gene in 10q24.32. Here, we report on an 18-month-old boy presenting with medulloblastoma, frontal bossing, and multiple skeletal anomalies and his father who has basal cell carcinomas, palmar pits, macrocephaly, bifid ribs, calcification of falx cerebri, and a history of surgery for odontogenic keratocyst. These clinical findings were compatible with the diagnosis of NBCCS, and a novel mutation, c.1249delC; p.Gln417Lysfs*15, was found in PTCH1 causing a premature stop codon.
Medulloblastoma (MB), the tumor of the cerebellum, is the most frequent brain cancer in childhood and a major cause of pediatric mortality. Based on gene profiling, four MB subgroups have been identified, i.e., Wnt or Sonic Hedgehog (Shh) types, and subgroup 3 or 4. The Shh-type MB has been shown to arise from the cerebellar precursors of granule neurons (GCPs), where a hyperactivation of the Shh pathway leads to their neoplastic transformation. We have previously shown that the gene Tis21 (PC3/Btg2) inhibits the proliferation and promotes the differentiation and migration of GCPs. Moreover, the overexpression or the deletion of Tis21 in Patched1 heterozygous mice, a model of spontaneous Shh-type MB, highly reduces or increases, respectively, the frequency of MB. Here we tested whether Tis21 can inhibit MB allografts. Athymic nude mice were subcutaneously grafted with MB cells explanted from Patched1 heterozygous mice. MB allografts were then injected with adeno-associated viruses either carrying Tis21 (AAV-Tis21) or empty (AAV-CBA). We observed that the treatment with AAV-Tis21 significantly inhibited the growth of tumor nodules, as judged by their volume, and reduced the number of proliferating tumor cells (labeled with Ki67 or BrdU), relative to AAV-CBA-treated control mice. In parallel, AAV-Tis21 increased significantly tumor cells labeled with early and late neural differentiation markers. Overall the results suggest that Tis21-gene therapy slows down MB tumor growth through inhibition of proliferation and enhancement of neural differentiation. These results validate Tis21 as a relevant target for MB therapy.
Cancer cells often express differentiation programs unrelated to their tissue of origin, although the contribution of these aberrant phenotypes to malignancy is poorly understood. An aggressive subgroup of medulloblastoma, a malignant pediatric brain tumor of the cerebellum, expresses a photoreceptor differentiation program normally expressed in the retina. We establish that two photoreceptor-specific transcription factors, NRL and CRX, are master regulators of this program and are required for tumor maintenance in this subgroup. Beyond photoreceptor lineage genes, we identify BCL-XL as a key transcriptional target of NRL and provide evidence substantiating anti-BCL therapy as a rational treatment opportunity for select MB patients. Our results highlight the utility of studying aberrant differentiation programs in cancer and their potential as selective therapeutic vulnerabilities.
Medulloblastoma (MB) is the most common malignant brain tumor in childhood. It contains at least four distinct molecular subgroups. The aim of this study is to explore novel diagnostic and potential therapeutic markers within each subgroup of MB, in particular within Group 4, the largest subgroup, to facilitate diagnosis together with gene therapy. One hundred and six MB samples were examined. Tumor subtype was evaluated with the NanoString assay. Several novel tumor related genes were shown to have high subgroup sensitivity and specificity, including PDGFRA, FGFR1, and ALK in the WNT group, CCND1 in the SHH group, and α-synuclein (SNCA) in Group 4. Knockdown and overexpression assays of SNCA revealed the ability of this gene to inhibit tumor invasion and induce apoptosis. Methylation-specific PCR and pyrosequencing analysis showed that epigenetic mechanisms, rather than DNA hypermethylation, might play the key role in the regulation of SNCA expression in MB tumors. In conclusion, we identify SNCA as a novel diagnostic biomarker for Group 4 MB. Some other subgroup signature genes have also been found as candidate therapeutic targets for this tumor.
BACKGROUND: Extraneural metastases are relatively rare manifestations of medulloblastoma.
CASE PRESENTATION: We present the case of a young boy with group three MYCN-amplified medulloblastoma. He received multimodal chemotherapy consisting of gross total resection followed by postoperative craniospinal radiation and adjuvant chemotherapy. The patient developed extraneural metastases 4 months after the end of therapy. Literature review identifies the poor prognosis of MYCN-amplified medulloblastomas as well as extraneural metastases; we review the current limitations and future directions of medulloblastoma treatment options.
CONCLUSION: To the best of our knowledge, this is the first molecularly characterized report of extraneural metastases of medulloblastoma in a child.
Chen YD, Zhang N, Qiu XG, et al.
LncRNA CDKN2BAS rs2157719 genetic variant contributes to medulloblastoma predisposition.J Gene Med. 2018; 20(1) [
PubMed]
Related Publications
BACKGROUND: How germline single nucleotide polymorphisms are involved in the etiology of medulloblastoma remans poorly understood. We hypothesized that CCDKN2A/B rs1063192 and rs4977756 and also the long noncoding RNA (lncRNA) CDKN2BAS rs2157719 glioma susceptibility polymorphisms identified by genome-wide association studies may contribute to medulloblastoma predisposition.
METHODS: To test this hypothesis, we genotyped these genetic variants among 160 medulloblastoma patients and 443 health controls in a Chinese population. Odds ratios (ORs) and 95% confidence intervals (CIs) were estimated by logistic regression.
RESULTS: We found that only the lncRNA CDKN2BAS rs2157719 T>C genetic polymorphism was significantly associated with an increased medulloblastoma risk (C allele: OR = 1.85, 95% CI = 1.32-2.58; p = 2.7 × 10
CONCLUSIONS: The findings of the present study provide important insights into the genetic complexities and predisposition of medulloblastoma in Chinese, especially at the lncRNA germline variation level.
Constitutional mismatch repair deficiency (CMMRD) is an autosomal recessively inherited childhood cancer susceptibility syndrome caused by biallelic germline mutations in one of the mismatch repair (MMR) genes. The spectrum of CMMRD-associated tumours is very broad and many CMMRD patients additionally display signposting non-neoplastic features, most frequently café-au-lait macules and other pigmentation alterations. We report on a 13-month-old girl suspected of having CMMRD due to a desmoplastic medulloblastoma and a striking skin pigmentation that included multiple café-au-lait macules, hypopigmented areas and Mongolian spots. Whole-exome sequencing revealed homozygosity for MSH2 variant p.(Leu92Val) and MSH6 variant p.(Val809del), both variants of uncertain significance (VUS). Immunohistochemical analysis of the tumour tissue showed expression of all four MMR proteins and gMSI testing was negative. However, functional assays demonstrated that the cells of the patient displayed methylation tolerance and ex vivo microsatellite instability, which unequivocally confirmed the diagnosis of CMMRD. Taken together, the results render the MSH2 variant unlikely to be responsible for the phenotype, while they are compatible with MSH6-associated CMMRD. This case illustrates the diagnostic strategy of confirming CMMRD syndrome in patients with VUS.
Medulloblastoma is the most common malignant childhood brain tumor. The heterogeneous tumors are classified into four subgroups based on transcription profiles. Recent developments in genome-wide sequencing techniques have rapidly advanced the understanding of these tumors. The high percentages of somatic alterations of genes encoding chromatin regulators in all subgroups suggest that epigenetic deregulation is a major driver of medulloblastoma. In this report, we review the current understanding of epigenetic regulation in medulloblastoma with a focus on the functional studies of chromatin regulators in the initiation and progression of specific subgroups of medulloblastoma. We also discuss the potential usage of epigenetic inhibitors for medulloblastoma treatment.
Zhang Y, Wang T, Wang S, et al.
Nkx2-2as Suppression Contributes to the Pathogenesis of Sonic Hedgehog Medulloblastoma.Cancer Res. 2018; 78(4):962-973 [
PubMed]
Related Publications
Aberrant Hedgehog signaling and excessive activation of the Gli family of transcriptional activators are key drivers of medulloblastoma (MB), the most common human pediatric brain malignancy. MB originates mainly from cerebellar granule neuron progenitors (CGNP), but the mechanisms underlying CGNP transformation remain largely obscure. In this study, we found that suppression of the noncoding RNA Nkx2-2as promoted Sonic Hedgehog (Shh)-potentiated MB development. Nkx2-2as functioned as a competing endogenous RNA against miR-103 and miR-107, sequestering them and thereby derepressing their tumor suppressive targets BTG2 and LATS1 and impeding cell division and migration. We also found that Nkx2-2as tethered miR-548m and abrogated its LATS2 targeting activity. Shh signaling impaired Nkx2-2as expression by upregulating the transcriptional repressor FoxD1. In clinical specimens of Shh-subgroup MB, we validated coordinated expression of the aforementioned proteins. Notably, exogenous expression of Nkx2-2as suppressed tumorigenesis and prolonged animal survival in MB mouse models. Our findings illuminate the role of noncoding RNAs in Hedgehog signaling and MB occurrence, with implications for identifying candidate therapeutic targets for MB treatment.
Further References
Medulloblastoma (MB) is the most common malignant brain tumor of children. To identify the genetic alterations in this tumor type, we searched for copy number alterations using high-density microarrays and sequenced all known protein-coding genes and microRNA genes using Sanger sequencing in a set of 22 MBs. We found that, on average, each tumor had 11 gene alterations, fewer by a factor of 5 to 10 than in the adult solid tumors that have been sequenced to date. In addition to alterations in the Hedgehog and Wnt pathways, our analysis led to the discovery of genes not previously known to be altered in MBs. Most notably, inactivating mutations of the histone-lysine N-methyltransferase genes MLL2 or MLL3 were identified in 16% of MB patients. These results demonstrate key differences between the genetic landscapes of adult and childhood cancers, highlight dysregulation of developmental pathways as an important mechanism underlying MBs, and identify a role for a specific type of histone methylation in human tumorigenesis.
Medulloblastoma is an aggressively growing tumour, arising in the cerebellum or medulla/brain stem. It is the most common malignant brain tumour in children, and shows tremendous biological and clinical heterogeneity. Despite recent treatment advances, approximately 40% of children experience tumour recurrence, and 30% will die from their disease. Those who survive often have a significantly reduced quality of life. Four tumour subgroups with distinct clinical, biological and genetic profiles are currently identified. WNT tumours, showing activated wingless pathway signalling, carry a favourable prognosis under current treatment regimens. SHH tumours show hedgehog pathway activation, and have an intermediate prognosis. Group 3 and 4 tumours are molecularly less well characterized, and also present the greatest clinical challenges. The full repertoire of genetic events driving this distinction, however, remains unclear. Here we describe an integrative deep-sequencing analysis of 125 tumour-normal pairs, conducted as part of the International Cancer Genome Consortium (ICGC) PedBrain Tumor Project. Tetraploidy was identified as a frequent early event in Group 3 and 4 tumours, and a positive correlation between patient age and mutation rate was observed. Several recurrent mutations were identified, both in known medulloblastoma-related genes (CTNNB1, PTCH1, MLL2, SMARCA4) and in genes not previously linked to this tumour (DDX3X, CTDNEP1, KDM6A, TBR1), often in subgroup-specific patterns. RNA sequencing confirmed these alterations, and revealed the expression of what are, to our knowledge, the first medulloblastoma fusion genes identified. Chromatin modifiers were frequently altered across all subgroups. These findings enhance our understanding of the genomic complexity and heterogeneity underlying medulloblastoma, and provide several potential targets for new therapeutics, especially for Group 3 and 4 patients.
Medulloblastomas are the most common malignant brain tumours in children. Identifying and understanding the genetic events that drive these tumours is critical for the development of more effective diagnostic, prognostic and therapeutic strategies. Recently, our group and others described distinct molecular subtypes of medulloblastoma on the basis of transcriptional and copy number profiles. Here we use whole-exome hybrid capture and deep sequencing to identify somatic mutations across the coding regions of 92 primary medulloblastoma/normal pairs. Overall, medulloblastomas have low mutation rates consistent with other paediatric tumours, with a median of 0.35 non-silent mutations per megabase. We identified twelve genes mutated at statistically significant frequencies, including previously known mutated genes in medulloblastoma such as CTNNB1, PTCH1, MLL2, SMARCA4 and TP53. Recurrent somatic mutations were newly identified in an RNA helicase gene, DDX3X, often concurrent with CTNNB1 mutations, and in the nuclear co-repressor (N-CoR) complex genes GPS2, BCOR and LDB1. We show that mutant DDX3X potentiates transactivation of a TCF promoter and enhances cell viability in combination with mutant, but not wild-type, β-catenin. Together, our study reveals the alteration of WNT, hedgehog, histone methyltransferase and now N-CoR pathways across medulloblastomas and within specific subtypes of this disease, and nominates the RNA helicase DDX3X as a component of pathogenic β-catenin signalling in medulloblastoma.
Recurring Structural Abnormalities
Selected list of common recurrent structural abnormalities
This is a highly selective list aiming to capture structural abnormalies which are frequesnt and/or significant in relation to diagnosis, prognosis, and/or characterising specific cancers. For a much more extensive list see the Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer.
Isochromosome 17q in Medulloblastoma
Biegel JA, Rorke LB, Packer RJ, et al.
Isochromosome 17q in primitive neuroectodermal tumors of the central nervous system.Genes Chromosomes Cancer. 1989; 1(2):139-47 [
PubMed]
Related Publications
We have prepared karyotypes from 22 primitive neuroectodermal tumors (PNETs) from pediatric patients ranging in age from 10 months to 16 years. Twenty-one cases were newly diagnosed, primary, posterior fossa tumors. One case was a recurrent tumor in a patient previously treated with radiation. Cytogenetic results were obtained from direct preparations and/or short-term (1-10 day) culture. Three tumors had apparently normal karyotypes. Nineteen tumors demonstrated numerical and/or structural abnormalities. The most frequent structural chromosomal changes were deletions and nonreciprocal translocations. Four tumors contained double minutes. Several chromosomes appear to be nonrandomly involved in PNETs. These include chromosomes 5, 6, 11, 16, 17, and a sex chromosome. The most consistent change, however, was an i(17q), present in one-third (8/22) of the cases. Strikingly, in three of these eight tumors, the i(17q) was the only structural abnormality observed. An i(17q) is not specific for pediatric PNETs, as it is also seen in leukemias and other solid tumors. However, in PNETs it may be a primary change related to tumor development and/or progression. Clinically, there was no correlation of the cytogenetic findings with histologic features of the tumors, size of the tumor, extent of metastasis, or surgical resection.
Giordana MT, Migheli A, Pavanelli E
Isochromosome 17q is a constant finding in medulloblastoma. An interphase cytogenetic study on tissue sections.Neuropathol Appl Neurobiol. 1998; 24(3):233-8 [
PubMed]
Related Publications
Isochromosome 17q (i[17q]) is the most frequent chromosomal abnormality in medulloblastoma, occurring in 30-60% of cases by karyotype analysis. In the present study i[17q] was demonstrated in routinely processed tissue sections of 20 medulloblastomas by in situ hybridization (ISH), using a chromosome 17 centromeric alpha satellite DNA probe. All medulloblastomas showed the i[17q] specific signal, i.e. two hybridization spots slightly apart from each other. The specific hybridization signal was not observed in ependymomas, cerebellar astrocytomas, haemangioblastomas, supratentorial neuroblastomas and ependymoblastomas. The constant finding of i[17q] in medulloblastoma depends on the much higher number of nuclei which can be analysed by ISH compared with cytogenetic techniques. Molecular data on medulloblastoma are consistent with the present results. The number of cells with i[17q] in medulloblastoma cases ranged from 3% to 9%; these figures are underestimated because of nuclear truncation in tissue sections. The percentage was not linked to patients' age, location of tumour, MIB-1 labelling index and histological type (classical vs desmoplastic). The present results indicate that i[17q] is a key event in the pathogenesis of medulloblastoma, and suggest a genetic difference between medulloblastoma and other primitive neuroectodermal tumours.
 Etoposide
Etoposide
